Nature Communications published on line a research paper entitled "Genomic Insights into Local Adaptation and Future Climate-Induced Vulnerability of a Keystone Forest Tree in East Asia" on November 1, 2022. The paper records the findings of a joint research project of three research groups led respectively by Professor Jing Wang, Professor Kangshan Mao and Professor Jianquan Liu from the College of Life Sciences of Sichuan University. This study took Populus koreana, an important tree species distributed in the temperate coniferous and broad-leaved mixed forest in Northeast China, as the research object, de novo and annotated the reference genome of Populus koreana at the chromosome level, and re sequenced the whole genome of 230 individuals from 24 natural populations of Populus koreana collected in the field.
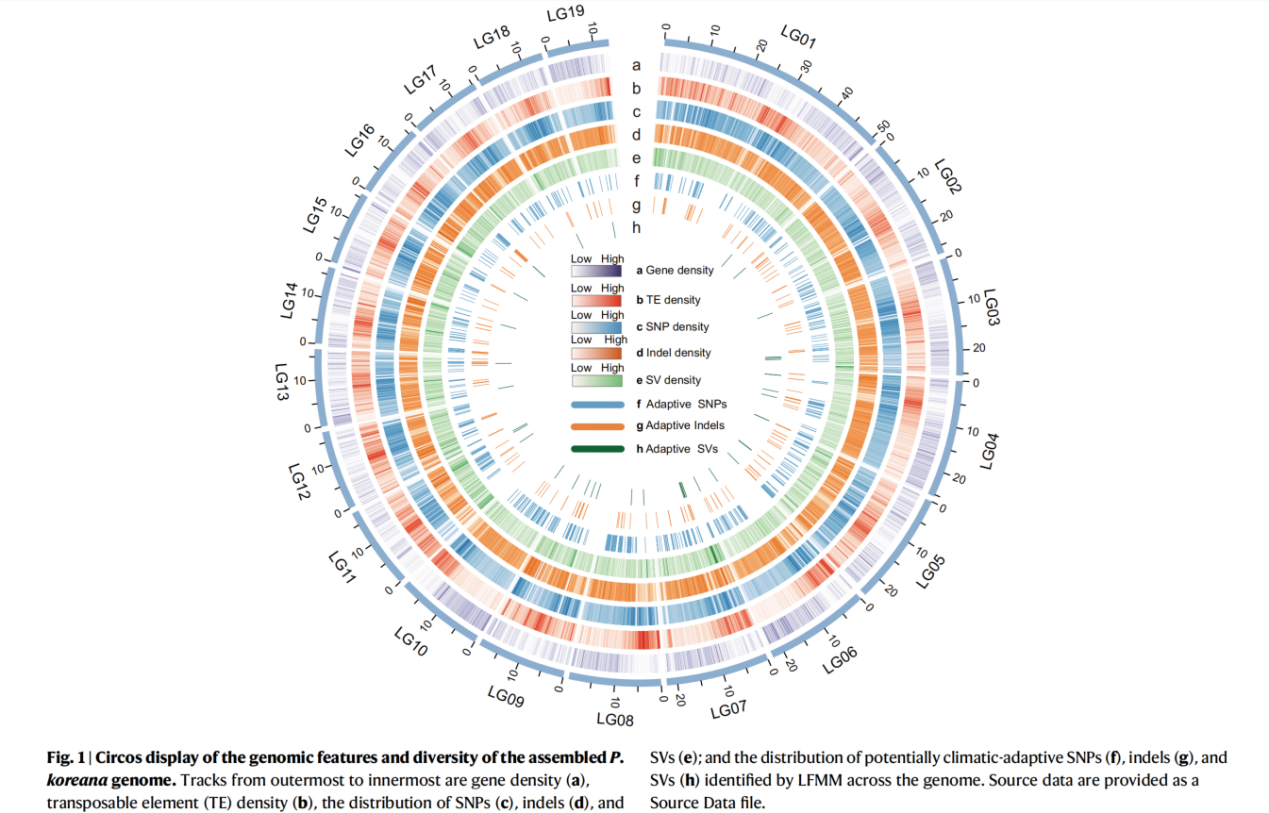
“For de novo assembly of the P. koreana genome, we integrated data from three sequencing and assembly technologies: ~42.42 Gb of Nanopore long-read sequencing (106×), ~29.82 Gb of short-read Illumina sequencing (74×), and ~54.22 Gb of Hi-C paired-end reads (137×) (Supplementary Tables 1–4). The final assembly captured 401.4 Mb of the genome sequence, with contig N50 of 6.41 Mb and ~99.6% (~399.94 Mb) of the contig sequences anchored to 19 pseudo-chromosomes (Fig. 1; Supplementary Fig. 1; Table 1; Supplementary Table 5). The high quality, continuity, and completeness of the assembled genome were supported by a high mapping rate (99.4%) of Illumina short reads and 97.8% of the single-copy orthologs from the Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis (Supplementary Table 6).” (Results)
“Rapid global climate change is posing a substantial threat to biodiversity. The assessment of population vulnerability and adaptive capacity under climate change is crucial for informing conservation and mitigation strategies. Here we generate a chromosome-scale genome assembly and re-sequence genomes of 230 individuals collected from 24 populations for Populus koreana, a pioneer and keystone tree species in temperate forests of East Asia. We integrate population genomics and environmental variables to reveal a set of climate-associated single-nucleotide polymorphisms, insertion/deletions and structural variations, especially numerous adaptive non-coding variants distributed across the genome. We incorporate these variants into an environmental modeling scheme to predict a highly spatiotemporal shift of this species in response to future climate change. We further identify the most vulnerable populations that need conservation priority and many candidate genes and variants that may be useful for forest tree breeding with special aims. Our findings highlight the importance of integrating genomic and environmental data to predict adaptive capacity of a key forest to rapid climate change in the future.” (Abstract)
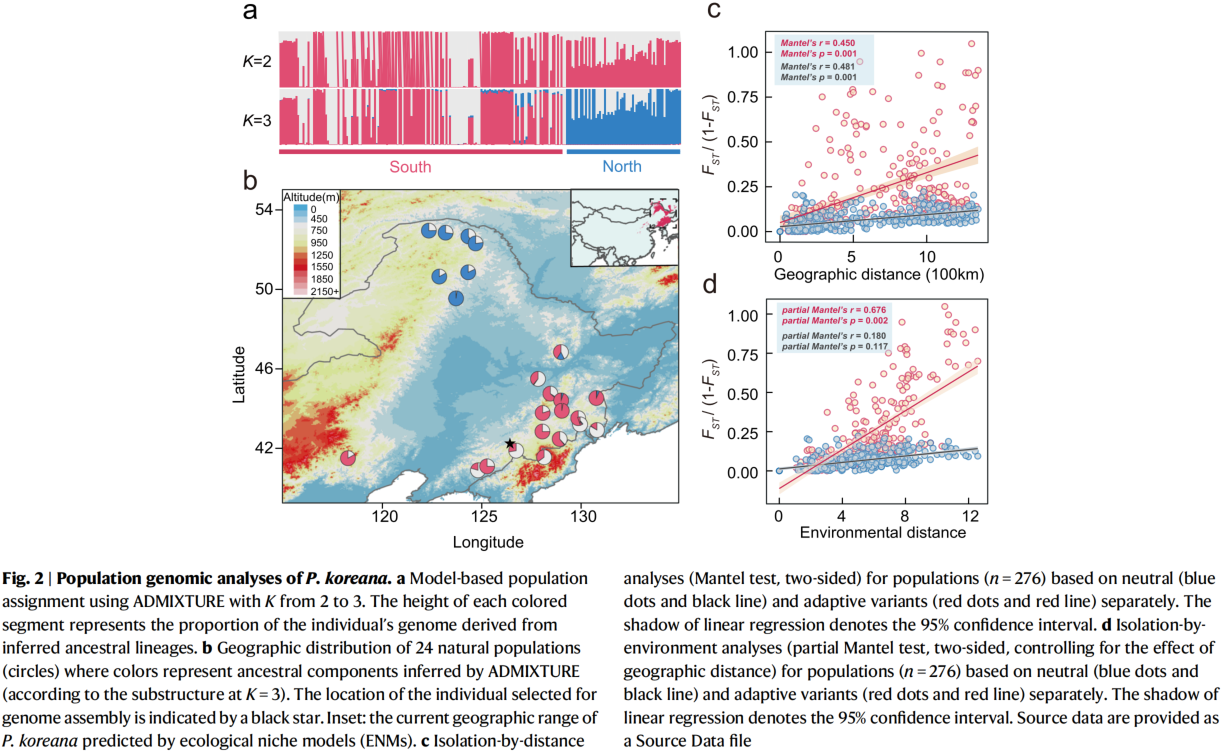
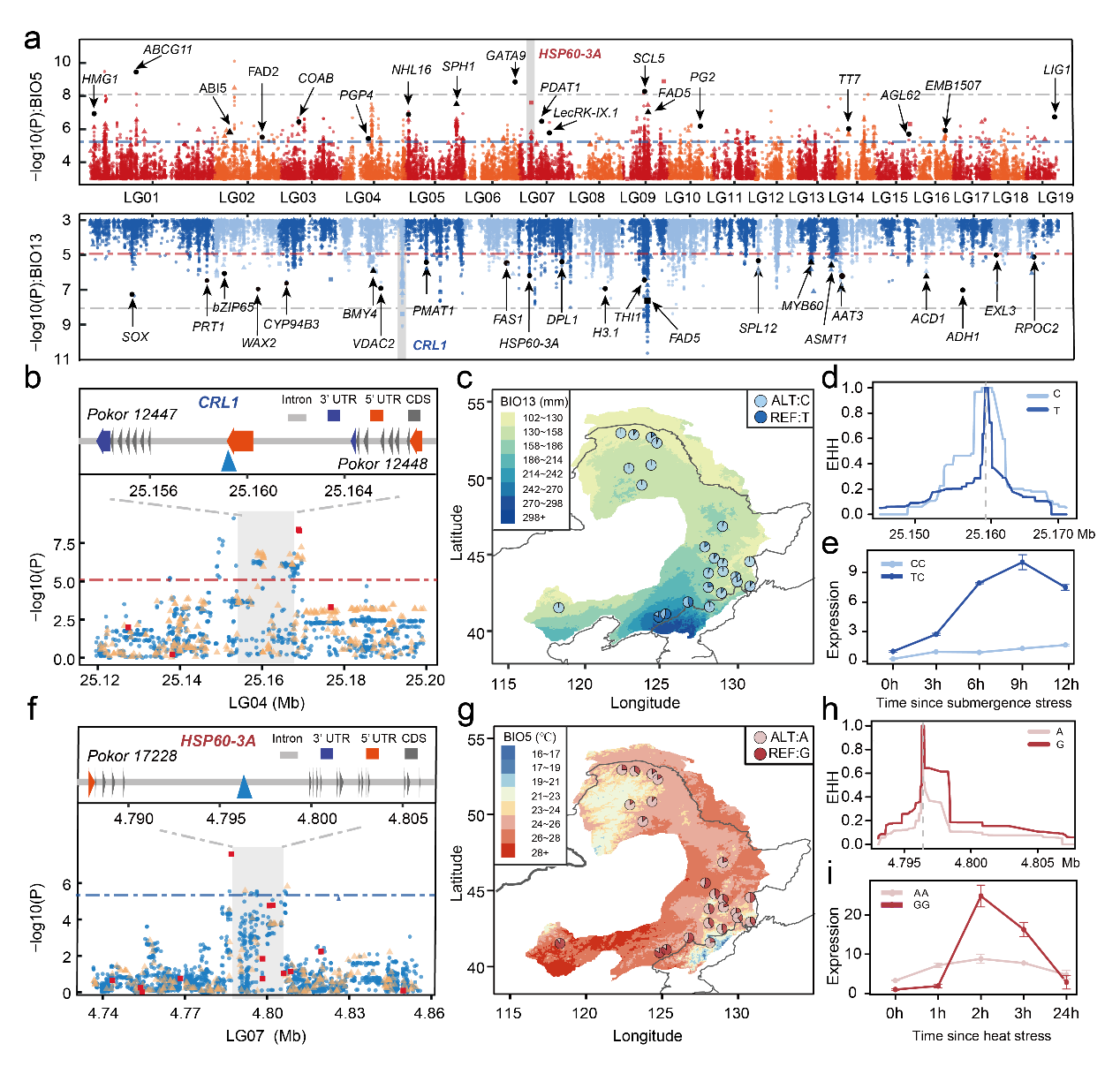

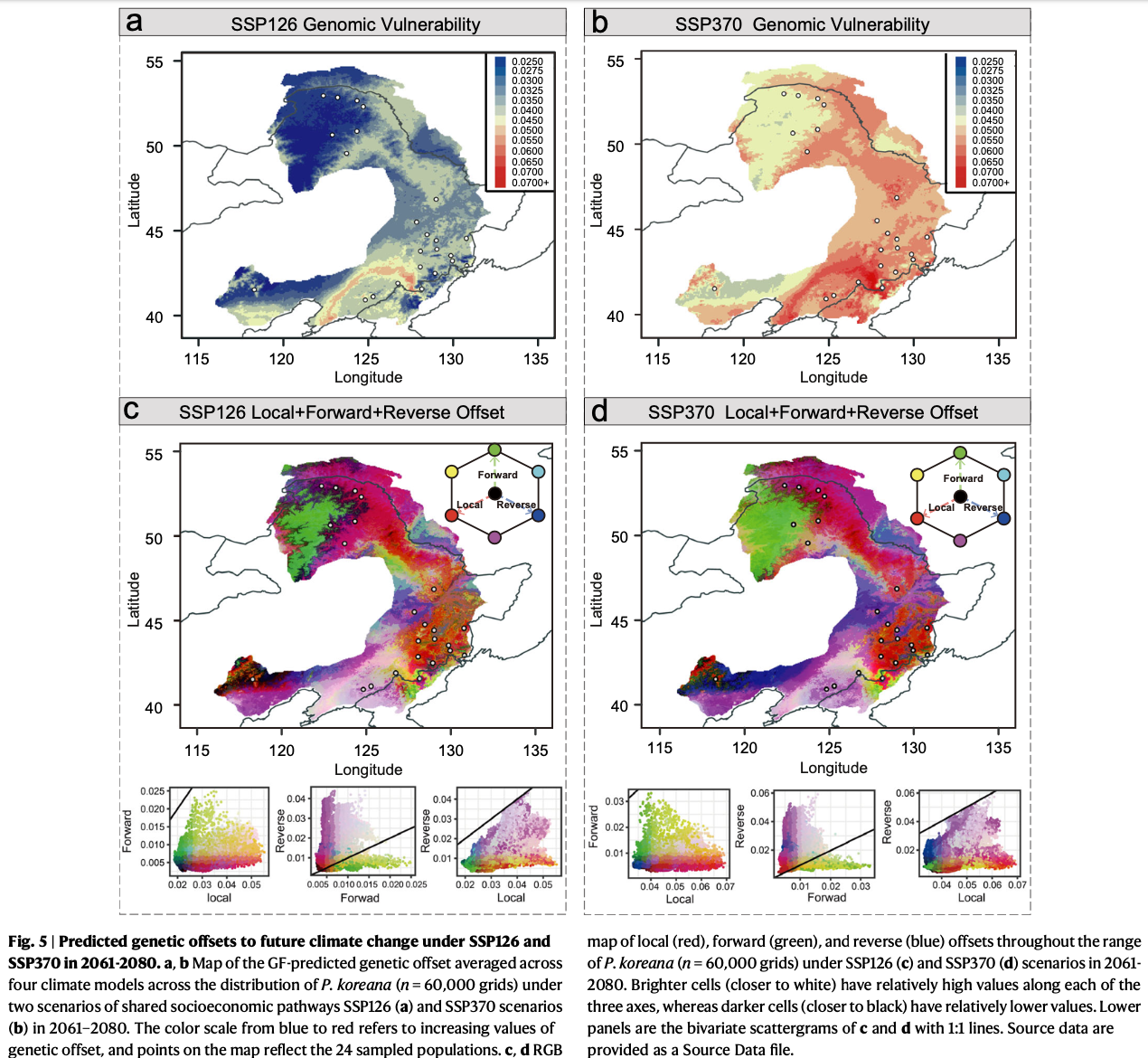
In this paper, 230 weight sequencing data from East Asia, combined with genomics and population genetics theory, were used to comprehensively explain the adaptability mode of poplar to the contemporary and future environment by innovative methods, providing important information for breeding and germplasm protection. At the same time, the study emphasized the importance of integrating genome and environmental data to predict the adaptive capacity of species to rapid climate change in the future, which has some inspiration for the study of adaptive evolution mechanism of forest tree species.
Jing Wang, Kangshan Mao and Jianquan Liu are the corresponding authors. Yupeng Sang, a doctoral student, and Zhiqin Long, a graduate student in Jing Wang's research group, are the co first authors of the paper. This work was supported by the National Key Research and Development Program of China (2022YFD2201200 to J.W., 2021YFD2200202 to J.L.), National Natural Science Foundation of China (31971567 to J.W.) and Fundamental Research Funds for the Central Universities (SCU2022D003, SCU2021D006, SCU2019D013 and 2020SCUNL207 to J.L., K.M., J.W.).
https://www.nature.com/articles/s41467-022-34206-8






